Beating Tox21 challenge with way too many parameters
Task: predict all 1.5k possible toxic effects of a given compound, given its name.
Solution: Deep Learning
Written by @tornikeo and @copilot
This is one of my favourite projects from 2021. So, naturally, I’d like everyone reading this to understand the task at hand as well as a glimpse into how my solution works ![]() .
.
Introduction
When a pharmaceutical company announces a groundbreaking new drug, usually people don’t think too hard about all the hard work and funding that went into developing that drug. What people do notice, however, is the price. New drugs cost a lot more than the generic over-the-shelf drugs, e.g aspirin. This is because the pharma has to cover the development expenses – including making sure that the drug is safe for human consumption.

That last part is what the Tox21 dataset is all about. As you might have already guessed, the dataset is about predicting the toxicity of new drugs. Whether or not the drugs actually do work is a whole ‘nother field, and Tox21 doesn’t touch that.

-- wikipedia

-- wikipedia
Usually, the toxicity tests are first done on tissue samples under the microscope, these being called in vitro tests, and then on some unlucky lab animals – called in vivo tests. Huge ethical problems aside, this process is very expensive. So, much so that many companies are now considering implementing a third kind of testing, aptly called in silico testing.
In silico tests (meaning in silicon, or in computer chip) are computer programs that take into account thousands of parameters about a given chemical in order to predict its toxicity.
“Robots are taking our jobs!”
– Unemployed laboratory rats (circa 2030)
And that’s where this project comes in. We have seen time and time again how neural networks can learn to solve complex problems. The goal here is to design a neural network that can accurately predict the result of chemical-to-cell interactions.
Now, what exactly does “chemical-to-cell interactions” mean? Well, this can be explained in a lot of ways, but, since I am no biochemist, I’ll try to stick to the AI-side of the things.
Interactions, a lot of them
The entire thing I am solving here can be represented as a neat, 3 column table, as shown below:
| Name of chemical | Name of toxic effect | Is toxic? |
|---|---|---|
| Aspirin | Cell death | YES |
| Aspirin | DNA damage | NO |
| Adrenaline | Cell death | NO |
| Adrenaline | DNA damage | YES |
In this grossly incorrect sample table, we have two chemicals (aspirin and adrenaline), two possible effects (cell death and DNA damage) and two possible results (YES and NO). The first two columns are given, the last column has to be predicted.
The problem is, most of these interactions are missing. In fact, out of the possible 13.9 million interactions, we only have access to 3.7 (a mere 25.5% coverage!).

Too many sparse features
Initially I was handed a dataset of interactions (described above) and another dataset for chemical descriptions. The latter was supposed to contain the information about the chemicals – in a machine-friendly format. As it turned out later, this was not at all helpful for the deep learning approach.
| index | V1 | V2 | V3 | V4 | V5 | V1071 | V1072 | V1073 | V1074 | V1075 |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 60-35-5 | 178 | 59.0371 | -0.808 | 43.09 | 0 | 0 | 0 | 0 | 0 |
| 1 | 103-90-2 | 1983 | 151.063 | 0.87 | 49.33 | 0 | 0 | 0 | 0 | 0 |
| … | ||||||||||
| 8829 | 541-85-5 | 7822 | 128.12 | 2.337 | 17.07 | 0 | 0 | 0 | 0 | 0 |
| 8830 | 61949-76-6 | 40326 | 390.079 | 6.13 | 35.53 | 0 | 0 | 0 | 0 | 0 |
The V1 to V1075 column names are not helpful at all. To make sense of these columns, a separate csv file was provided, features_id_name_mappings.csv. The file contains readable descriptions for what the columns describe.
| ID | DESC |
|---|---|
| FeaID | FeaName |
| V1 | casn |
| V2 | PubChem_CID |
| … | |
| V1074 | ClC1C(Br)CCC1 |
| V1075 | BrC1C(Br)CCC1 |
So, now, we have yet another problem to tackle - the dataset is sparse and requires special handling to work with.
So what do we do?
Two words: Embeddings, and Masking.
The issue with sparse data is apparent - neural networks need a huge amount of compute to crunch down the truly enormous - and mostly empty - vectors into the hidden dense representations. In our case, using a simple linear layer would have to have a shape of \(O(M_{sparse} N_{hidden})\). With \(N_{hidden}\) being at least 100, this nets us a \(1000 \times 100\) elements just for the first layer alone.
Luckily, a 2017 paper, ‘Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition’ provided just the right tools for fixing this issue. The Mol2Vec authors trained a large Node2Vec-style model on raw molecular graph data. My blog on the “DeepWalk: Online Learning of Social Representations:” explains a related, but slightly different method of how to train neural networks on graph data. The core concept is still the same - Create an auto-encoder style network and train it to predict the graph patterns. After the network converges, use the encoder part of the network as a graph-to-dense transformation tool: you input raw graph data - out comes an \(N\)-dimensional vector that contains a useful representation of the input. In contrast to the classical fingerprinting approaches, this approach has two major benefits:
- It is much less biased (i.e. it only depends on the data and, as such, is less susceptible to human error).
- Requires much less compute due to dense representation (300 “float32” entries, vs 1100 sparse “float32” entries.)
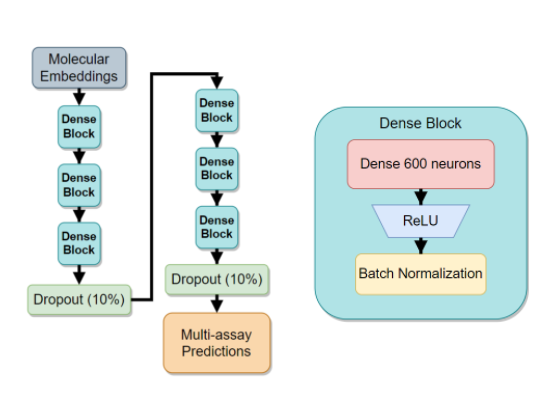
The second word, masking, refers to our unique solution to the overwhelming amount of missing data, along with the need assay encoding. The latter problem of encoding assays comes from the fact that there are over 1500 different assays and we need to somehow input all 1500 into the network with as little computational burden as possible. Recall also, that we are given a very long list of chemical-assay pairs and a corresponding binary result active or inactive. So, how do we solve all these issues with a single modification?
TODO: Show pivot matrix here
It’s simple:
- Create a pivot matrix - Chemicals go into rows, assays go into columns.
- Each intersection of the pivot matrix is either a 0 or a 1, corresponding to the inactive and active labels.
- To encode missing values, create a separate binary matrix of the same size, where 0 refers to a missing chemical-assay interaction, and 1 otherwise.
There you have it. Assay encoding is taken care of, since each column will be predicted by a separate output neuron (and GPU acceleration makes training a breeze). But what about the missing data?
Well, for that, we modify the binary cross-entropy loss function.
To be continued …